Dedno stanje SMA je posledica mutacij v genu SMN1 ali genu SMN2, katerega namen je proizvesti beljakovino, ki služi preživetju motoričnih nevronov.
Obstaja pet različnih oblik spinalne mišične atrofije: tip 0, tip 1, tip 2, tip 3 in tip 4. Prve tri vrste so zelo resne in povzročijo, da bolnik prezgodaj umre; tipa 3 in tip 4 sta blažji različici, ki vplivata na življenjski standard bolnika, vendar ne povzročata prezgodnje smrti.
Za diagnozo SMA je potreben genetski test na vzorcu krvi.
Trenutno zdravljenje SMA temelji predvsem na simptomatskem zdravljenju, katerega cilj je lajšanje motenj in obvladovanje zapletov. Na voljo je zdravilo, ki temelji na načelih genske terapije, vendar je zelo draga rešitev in velja le za določene bolnike.
, ki se kaže z atrofijo in posledično oslabitvijo skeletnih mišic ter motoričnimi težavami.
SMA je stanje, ki lahko povzroči smrt bolnika v mladosti ali zelo mladosti: najresnejše oblike bolezni dejansko vplivajo na učinkovitost dihalnih mišic in so odgovorne za epizode odpovedi dihanja ali pljučnico z usoden izid.
Motorni nevroni in SMA

Motorni nevroni ali motorični nevroni so živčne celice, ki nastanejo v centralnem živčnem sistemu (možgani in hrbtenjača) in ki s svojimi podaljški (aksoni) nadzorujejo aktivnost mišic in žlez.
Obstajata dve vrsti motoričnih nevronov: zgornji motorični nevroni (ali prvi motorični nevroni) in spodnji motorični nevroni (ali drugi motorični nevroni).
Zgornji motorični nevroni izvirajo iz možganov in usmerjajo aktivnost spodnjih motoričnih nevronov, ki nastanejo predvsem v hrbtenjači in so odgovorni za usmerjanje aktivnosti skeletnih (ali somatskih) mišic, gladkih (ali visceralnih) mišic, srčne mišice in srca.
Motorni nevroni posameznikov s SMA postopoma degenerirajo, kar povzroči "mišično atrofijo zaradi neaktivnosti, ki v najhujših primerih povzroči paralizo, odpoved dihanja in smrt.
Epidemiologija: kako pogosta je mišična atrofija hrbtenice?
SMA ima "letno pojavnost 1 primera na 10.000 novorojenčkov.
5 in od katerih je odvisna proizvodnja tako imenovanega proteina preživetja motoričnih nevronov (SMN).Kot kaže ime proteina, ki ga proizvajata SMN1 in SMN2, mutacija teh genov motornim nevronom odvzame biološko snov, ki je bistvena za njihovo preživetje; natančneje, zmanjšuje raven beljakovin: na primer v prisotnosti mutacij v SMN1 se raven beljakovin SMN zniža na 10-20% normalne.
Očitno je, da odsotnost ustreznih količin proteina SMN določa progresivno degeneracijo motoričnih nevronov.
Izguba motoričnih nevronov prekine živčno signalizacijo, ki omogoča nadzor aktivnosti mišic človeškega telesa; slednji se zaradi dejstva, da niso več uporabni, postopoma atrofirajo in oslabijo.
Ali ste vedeli, da ...
Gen SMN2 je za SMA gen, ki spreminja bolezen; pravzaprav se pri bolnikih z mutacijo v SMN1 in ki imajo iz nekega razloga tri ali štiri kopije gena SMN2 SMA pojavi v blažji obliki.
Spinalna mišična atrofija: Vrste mutacij
Kadar je SMA posledica "spremembe SMN1", je v 95-98% primerov odgovorna mutacija izbrisana celotnega gena, le v 2-5% pa "anomalija normalnega genskega zaporedja".
Spinalna mišična atrofija: dedna bolezen
V skoraj vseh primerih (98%) je genetska anomalija, odgovorna za SMA, dedna, torej jo prenašajo starši bolnega posameznika.
2% nedednih primerov SMA je posledica mutacije de novo se je zgodil v zelo zgodnji fazi embrionalnega razvoja.
SMA in model dedovanja
Dedni model za spinalno mišično atrofijo je avtosomno recesiven, kar pomeni, da sta za podedovanje SMA bistvena, da sta oba starša zdrava nosilca genetske okvare v SMN1 ali SMN2 in da jo preneseta oba starša.
V primeru avtosomno recesivnih dednih bolezni, kot je SMA, je verjetnost, da bosta oba zdrava prenašalca na otroka prenesla genetsko napako in tako zbolela, 25%ali ena na vsake 4 primere.
Vrste SMA
Na podlagi starosti nastopa in resnosti stanja strokovnjaki prepoznajo pet različnih oblik spinalne mišične atrofije:
- SMA tip 0: to je najhujša oblika od vseh. Pojavi se že pred rojstvom z zmanjšano gibljivostjo ploda.
Dojenčki običajno preživijo nekaj tednov po rojstvu, tudi če dobijo podporo za dihanje. - SMA tip 1: od oblik, ki se pojavijo v življenju, je najhujša in najpogostejša (približno 50% primerov); pojavi se v zgodnji starosti, običajno v šestem mesecu življenja.
Praviloma je vzrok smrti že v prvih letih življenja; redko, v adolescenci.
Smrt običajno nastopi zaradi "dihalne odpovedi" ali "okužbe pljuč". - SMA tip 2: je oblika, ki se po gravitaciji uvršča na drugo mesto; na splošno se začne med 7 in 18 meseci življenja.
Pričakovana življenjska doba prizadetih je večja kot v prejšnjem primeru: bolniki dejansko uspejo doseči odraslost. - SMA tip 3: manj huda kot prejšnja dva, ta oblika SMA se običajno pojavi po 18 mesecih življenja (v nekaterih primerih se lahko pojavi tudi v otroštvu ali adolescenci).
Vključuje velike invalidnosti, vendar ne vpliva na pričakovano življenjsko dobo. - SMA tip 4: je oblika bolezni pri odraslih in najmanj huda; običajno se začne okoli tretjega desetletja življenja in poteka zelo počasi.
Na splošno ni odgovoren za težave z dihanjem in je povezan z "normalno pričakovano življenjsko dobo".
Ravni beljakovin SMN vplivajo na resnost SMA: nižja je količina SMN, večja je resnost povezane bolezni.
Zmanjšanje ravni SMN je tesno povezano z obsegom genetske okvare, ki je prizadela gene SMN1 ali SMN2: bolj kot je ta okvara obsežnejša, bolj pomembno je zmanjšanje količine proteina SMN (to velja za na primer izbris gena).
Poleg tega SMA ne ogroža intelektualnih funkcij (IQ bolnikov je normalen) in prihrani vid.
Za dodatne informacije: SMA: vsi simptomiSimptomi SMA tipa 0
Kot smo že povedali, se SMA tipa 0 pojavlja že v prenatalni starosti z zmanjšano gibljivostjo ploda; ob rojstvu ima bolnik očitne težave pri požiranju in dihanju.
Bolezen povzroči smrt v nekaj tednih po rojstvu, tudi če bolnik dobi dihalno podporo.
Simptomi SMA tipa 1
Otroci s SMA tipa 1 imajo zelo šibke mišice, ki se ne razvijajo tako, kot bi morale (izguba mišic). To jim onemogoča opravljanje dejavnosti, kot so dvigovanje glave, premikanje udov in prevzem sedečega položaja; poleg tega postopoma otežuje vitalne funkcije, kot so sesanje mleka, požiranje, žvečenje in dihanje.
Značilno je, da je SMA tipa 1 v prvih nekaj letih življenja usodna; nekateri pacienti pa uspejo doseči starost mladostnika.
Smrt običajno nastopi zaradi odpovedi dihanja ali "okužbe pljuč zaradi težav pri požiranju (pljučnica pri zaužitju ali pljučnica) ab ingestis).
Simptomi SMA tipa 2
SMA tip 2 se klasično kaže z:
- Mehkoba mišic rok in nog;
- Tresenje v prstih in rokah;
- Težave pri samostojnem prevzemanju sedečega položaja (pacient pa ga uspe ohraniti);
- Težave pri hoji in stanju
- Deformacija in težave s sklepi;
- Težave pri dihanju in požiranju hrane;
- Skolioza (običajno se pojavi kasneje).
Tudi v tem primeru so težave z dihanjem in požiranjem hrane vzrok za prezgodnjo smrt, ki se običajno pojavi na začetku odrasle dobe.
Simptomi SMA tipa 3
SMA tipa 3 povzroča težave z držo in ravnotežjem, tresenje rok in težave pri vstajanju iz sedečega položaja, hoji, plezanju po stopnicah in teku.
Bolezni na začetku ne potrebujejo podpore za gibanje; pozneje z degeneracijo večjega števila motoričnih nevronov postanejo bergle, sprehajalci in invalidski vozički temeljni.
Čeprav se to lahko zgodi, je zelo redko, da imajo bolniki s SMA tipa 3 težave z dihanjem in požiranjem hrane.
Ob prisotnosti te oblike SMA je pričakovana življenjska doba normalna, vendar z vsemi omenjenimi težavami.
Simptomi SMA tipa 4
Z nastopom odraslih je SMA tip 4 običajno povezan z:
- Oslabitev mišičnega tonusa v rokah in nogah;
- Težave pri hoji
- Tresenje in nenadno trzanje mišic.
Sprva so bile omenjene pritožbe zmerne; v starosti postanejo doslednejši.
Tako kot SMA tipa 3 tudi SMA tipa 4 ni bolezen, ki bi vplivala na bolnikovo pričakovano življenjsko dobo.
SMA: kdaj k zdravniku?
Vsem staršem, ki vedo, da so zdravi prenašalci SMA, toplo svetujemo, da se posvetujejo s pediatrom, ki ima izkušnje z genetskimi boleznimi, in genetikom.
Če nimate tovrstnih podatkov, je dobro iz meseca v mesec oceniti motorični razvoj vašega otroka in funkcije, od katerih je odvisno življenje (npr. Dihanje).
Vsekakor so nezmožnost sedenja ali zavzemanja sedečega položaja, težave pri hranjenju, pomanjkanje dihanja ter tanka in manj napeta muskulatura kot pri vrstnikih zvon za alarm.
Kar zadeva SMA za odrasle, obstaja sum na bolj ali manj nenaden pojav mišične oslabelosti in težave pri hoji, ki ga je treba spremljati.
Spinalna mišična atrofija: zapleti
Najhujše oblike SMA lahko povzročijo zaplete, kot so:
- Dušenje zaradi hrane. To je posledica zmanjšane sposobnosti žvečenja in zaužitja hrane.
- Odpoved dihanja. To je posledica nezmožnosti nadzora aktivnosti dihalnih mišic.
- Pljučnica ab ingestis (ali inhalacijska pljučnica). Pojavi se, ko tuj material, ki prenaša patogene, na primer hrana, slina ali nosni izločki, vstopi ali se nabere v pljučih.
Pljučnica ab ingestis je posledica težav pri požiranju. - Paraliza, ki je povzročila uporabo invalidskih vozičkov. To se zgodi, ko je bolezen nepopravljivo ogrozila bolnikove gibalne sposobnosti.
- Podhranjenost. To je še ena posledica težav pri požiranju: bolnik se v resnici trudi pravilno hraniti.
Opozoriti je treba, da se včasih pri diagnozi SMA lahko uporabijo testi, kot sta elektromiografija ali mišična biopsija.
SMA: Fizični pregled in anamneza
Fizični pregled pri pacientu, ki ima lahko SMA, vključuje natančno analizo simptomov in iskanje nekaterih značilnih znakov bolezni, kot so:
- Šibkost in občutljivost mišic;
- Nenadna krčenja mišic
- Zmanjšani ali odsotni tetivni refleksi.
Kar zadeva zdravstveno anamnezo, se to osredotoča predvsem na bolnikovo družinsko anamnezo, da bi ugotovili, ali se kateri od družinskih članov (starši, bratje in sestre, stari starši) pritožuje ali se pritožuje zaradi podobne simptomatologije. Očitno dejstvo, da je SMA dedna bolezen, ki se prenaša od staršev.
Čeprav ne dopuščajo dokončne diagnoze, lahko fizični pregled in anamneza prineseta zelo koristne informacije, ki usmerjajo preiskave k izvajanju genetskega testa.
Jasno je, da bodo starši, če je bolnik majhen otrok, med anamnezo sodelovali z zdravnikom.
SMA in genetski test

Genetski test za odkrivanje SMA vključuje iskanje in preučevanje mutacij v genih SMN1 / SMN2 v vzorcu krvnih celic bolnika.
Prisotnost genetskih sprememb očitno pomeni bolezen.
Analiza zaznanih mutacij je bistvena za določitev vrste prisotne spinalne mišične atrofije in resnosti stanja.
Za poznavanje rezultatov prej omenjenega genetskega testa je na splošno treba počakati od 3 do 4 tedne (natančne čakalne dobe se razlikujejo glede na genetski center, ki izvaja test).
SMA: ali je možna prenatalna diagnoza?
SMA je mogoče diagnosticirati v prenatalni starosti.
Če želite to narediti, potrebujete genetski test na vzorcu fetalnih celic, pridobljen z občutljivimi metodami, kot sta vilocenteza ali amniocenteza.
Glede na nevarnost splava, ki je značilna za CVS in amniocentezo, zdravniki opravljajo prenatalne raziskave za vse mutacije, ki jih je mogoče pripisati "spinalni mišični atrofiji", le če je za tem družinska anamneza SMA ali če je nerojeni otrok otrok zdravih nosilcev bolezni.
SMA in pregled novorojenčkov
Treba je omeniti, da je v nekaj italijanskih regijah (Lazio in Toskana) storitev aktivna presejanje za zgodnjo diagnozo SMA in drugih resnih genetskih bolezni.
Zgodnja diagnoza teh bolezni omogoča pravočasno načrtovanje najprimernejše simptomatske terapije za obvladovanje simptomov in zapletov.
Spinalna mišična atrofija in načrtovanje nosečnosti
Genetsko svetovanje je priporočljivo za vse ženske, ki iščejo nosečnost in imajo:
- V prejšnji nosečnosti sta imela otroka z SMA;
- Za seboj imajo družinsko anamnezo SMA;
- Ali so zdravi nosilci bolezni ali je njihov partner.
Genetsko svetovanje lahko ženskam s temi stanji pomaga razumeti, kakšnim tveganjem je izpostavljen bodoči otrok.
SMA in diferencialna diagnoza
Obstajata dve patologiji, ki sta zelo podobni SMA, ki le "temeljita diagnostična preiskava prepozna in prepreči zamenjavo s" spinalno mišično atrofijo: to sta "spinalna mišična atrofija z dihalno stisko (SMARD) in" bulbo-spinalna mišična atrofija (BSMA). Kennedyjeva bolezen); prvi je posledica mutacije gena IGHMBP2, ki se nahaja na 11. kromosomu, drugi pa zaradi mutacije spolnega kromosoma X.
in farmacevtski izdelki) odobrila zdravilo Zolgensma, metodo genske terapije za zdravljenje spinalne mišične atrofije.
Zolgensma je sestavljena iz zelo napredne tehnike molekularne biologije, ki vključuje uporabo virusnega vektorja, ki lahko vstavi normalno kopijo gena SMN1 / SMN2 v DNK, prisotno v motoričnih nevronih bolnika.
Dajanje prej omenjenega vektorja virusa poteka z intravensko injekcijo.
Zolgensma se je izkazala za učinkovito. Vendar pa ima po pričakovanjih dve glavni omejitvi, ki preprečujeta njeno skupno uporabo:
- Je zelo drago. Govori se o milijonih evrov;
- Uporablja se samo za bolnike z SMA, mlajše od 2 let.
Spinalna mišična atrofija: simptomatsko zdravljenje
Simptomatske terapije za SMA zagotavljajo večje koristi, če jih sprejmemo takoj; zato je zgodnja diagnoza bolezni zelo pomembna.
SMA in respiratorna podpora
Pravilna dihalna podpora bolnikom s SMA pomaga ne le dihati, ampak tudi zmanjša tveganje za pljučne okužbe.
Med različnimi terapevtskimi možnostmi obstajajo maske za neinvazivno prezračevanje in bolj invazivne rešitve, kot sta orotrahealna intubacija in traheostomija; prve so idealne za manj hude primere, medtem ko so bolj invazivne rešitve bistvene za bolnike z resnimi težavami.
Podpora SMA in prehrana
Najhujše oblike spinalne mišične atrofije vplivajo na sposobnost požiranja in žvečenja hrane, pri čemer je bolnik izpostavljen tveganju zadušitve, pljučne pljučnice in podhranjenosti.
Za obvladovanje teh nevarnih posledic je nujno, da se zatečemo k pripomočkom za hranjenje, kot sta nazogastrična sonda ali operacija gastrostomije, in se zanesemo na nutricionista, ki bo načrtoval prehrano, ki bo ustrezala pacientovim potrebam.
SMA in fizioterapija
Motorne težave, ki so značilne za bolnika s spinalno mišično atrofijo, vodijo do otrplosti sklepov in mišic zaradi neaktivnosti.
Ustrezen fizioterapevtski program vam omogoča, da kolikor je mogoče izboljšate prožnost mišic in sklepe naredite manj toge.
Jasno je, da ta program vključuje vaje, katerih izvajanje je v dosegu bolnikovih sposobnosti.
SMA in ortopedija
Ob prisotnosti skolioze, značilne za hude oblike SMA, se je nujno posvetovati z ortopedom; slednje bi lahko nakazovalo na uporabo ortopedskega steznika, če je deformacija blaga, ali na odločitev za operacijo spinalne fuzije, če je malformacija hrbtenice huda.
Zdravila proti SMA
Že nekaj let obstajajo tudi posebna zdravila proti SMA.
Ta zdravila si zaslužijo ločeno zdravljenje v primerjavi s simptomatsko terapijo, čeprav ne dovoljujejo ozdravitve bolezni, ampak le njeno zadrževanje.
Specifična zdravila proti SMA, ki so trenutno na voljo, sta Spinraza (nusinersen) in Evrysdi (risdiplam): prva delujeta tako, da v procesu popravljata aberantno proizvodnjo beljakovin SMN; druga povečujeta raven proizvodnje SMN in ju poskušata ohraniti na kvota, ki ustreza potrebam človeškega organizma.
Spinraza in Evrysdi sta leta 2017 odobrila FDA, rezultati pa v nekaterih primerih celo več kot zadovoljivi, vendar imajo pomembno omejitev: so zelo dragi.
Za dodatne informacije: Spinraza: Kako deluje, tveganja in koristi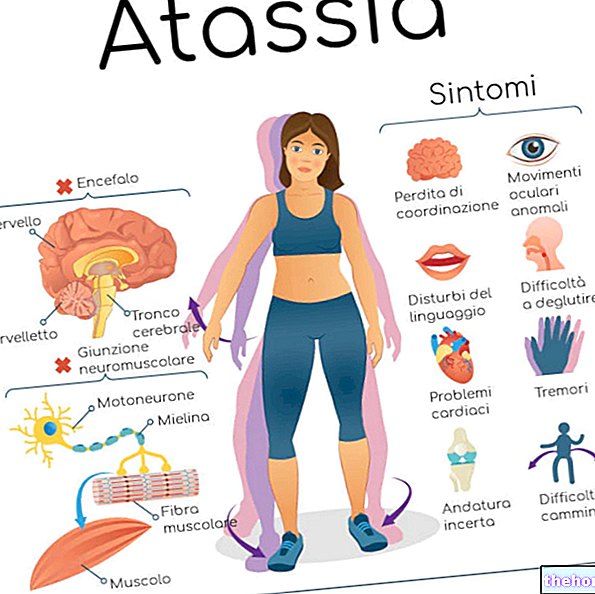









-non-sentire-gli-odori.jpg)