Splošnost
Osteogeneza imperfecta je prirojena genetska bolezen, ki ni povezana s spolom, odgovorna za določeno krhkost kosti in izrazito nagnjenost k zlomom.
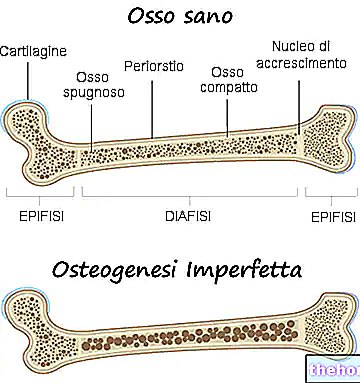
Simptomi osteogeneze imperfecta so številni; na splošno so sestavljeni iz: oslabitve kosti, velike nagnjenosti k zlomom kosti, prisotnosti modrih, sivih ali vijoličnih očesnih beločnic, prisotnosti deformacij kosti ali drugih skeletnih sprememb, trikotnega obraza, krhkosti zob itd. .
Na splošno so za pravilno diagnozo nepopolne osteogeneze bistveno naslednje: fizični pregled, anamneza, preskusi slikanja z medicinsko sliko, test za ocenjevanje kolagena tipa I in genetski test.
Žal so trenutno edino zdravljenje, ki je na voljo bolnikom z nepopolno osteogenezo, simptomatsko. Zadevna bolezen je pravzaprav neozdravljiva.
Kaj je osteogeneza imperfecta?
Osteogenesis imperfecta je genetska bolezen, zaradi katere so kosti prizadete osebe šibkejše in bolj nagnjene k zlomom.
V resnici se z izrazom osteogeneza imperfecta zdravniki nanašajo na heterogeno skupino genetskih bolezni, za katero je značilna določena stopnja krhkosti kosti. Obstaja torej več oblik (ali vrst) nepopolne osteogeneze, nekatere so veliko hujše od drugih.
TO JE PRIRODENA BOLEZEN
Pri ljudeh, ki jih ta bolezen prizadene, je osteogenesis imperfecta bolezen, ki je prisotna že od rojstva, zato jo je mogoče v vseh pogledih opredeliti kot prirojeno bolezen.
ALI JE POVEZAN S SPOLOM?
Osteogeneza imperfecta ni genetsko povezana s spolom, na primer hemofilija ali Klinefelterjev sindrom.
EPIDEMILOGIJA
Po nekaterih statističnih raziskavah bi bila incidenca nepopolne osteogeneze enaka enemu primeru na vsakih 15.000 do 20.000 rojstev. To pomeni, da ima vsakih 15.000–20.000 novorojenčkov enega, ki je prizadet zaradi nepopolne osteogeneze.
Druge statistične študije so pokazale tudi, da osteogeneza imperfecta enako vpliva na moške in ženske in da nima nobene naklonjenosti za določeno populacijo ali etnično skupino.
Življenjska doba je izjemno spremenljiv parameter, ki je odvisen od oblike nepopolne osteogeneze.
Vzroki
Pomanjkljiva osteogeneza je skoraj vedno posledica kvalitativne in kvantitativne spremembe proizvodnje kolagena tipa I.
Kolagen tipa I je bistven za krepitev kosti in za ohranjanje zdravega vezivnega tkiva, ki sestavljajo hrustanec, kite, kožo, očesno beločnico itd.
Zato sprememba v proizvodnji kolagena tipa I vpliva na trdnost kosti in dobro zdravje vezivnega tkiva v človeškem telesu.
KAJ SPREMENI PROIZVODNJO KOLAGENA?
Genetska bolezen je stanje, ki nastane zaradi mutacije enega ali več genov, ki sestavljajo celično DNK.
V primeru nepopolne osteogeneze je treba vzroke za slednje skoraj vedno najti v mutaciji enega ali obeh genov COL1A1 (na kromosomu 17) in COL1A2 (na kromosomu 7).
V normalnih pogojih COL1A1 in COL1A2 uravnavata normalno proizvodnjo kolagena tipa I; ob prisotnosti mutacij v svojem naboju ne uspejo pri regulacijski funkciji.
Pomembno: kateri drugi geni, če so mutirani, povzročajo nepopolno osteogenezo?
Poleg mutacij COL1A1 in COL1A2 so mutacije v genih IFITM5, SERPINF1, CRTAP in LEPRE1 potencialni vzroki za nepopolno osteogenezo.
Omenjeni geni zajemajo funkcije, ki se razlikujejo od COL1A1 in COL1A2 - zato ne nadzorujejo proizvodnje kolagena tipa I - vendar še vedno "vplivajo na moč in odpornost kosti človeškega okostja".
KAKŠNA JE GENETSKA BOLEZEN?
Osteogeneza imperfecta je avtosomna genetska bolezen.
Izraz avtosom, povezan z genetsko boleznijo, kaže, da je zadevno stanje posledica genetskih mutacij, ki temeljijo na avtosomnih in nespolnih kromosomih.
Bralce opozarjamo, da ima človek kromosomski niz 23 parov celotnih kromosomov, v katerih je 22 parov avtosomnega tipa in samo en par spolnega tipa. Par kromosomov spolnega tipa vpliva na spol posameznik.
Pomanjkljiva osteogeneza po mutacijah v COL1A1, COL1A2 in IFITM5 ima vse značilnosti avtosomno dominantne bolezni, kadar je posledica mutacij v genih SERPINF1, CRTAP in LEPRE1, ima značilnosti avtosomno recesivne bolezni.
VRSTE
Trenutno zdravniki verjamejo, da obstaja 8 vrst (ali oblik) osteogeneze imperfecta. Za razlikovanje med različnimi vrstami so se odločili uporabiti rimsko oštevilčenje, natančneje prvih osem rimskih številk.
Spodnja tabela prikazuje 8 oblik nepopolne osteogeneze, mutacije, ki jih povzročajo, in druge značilnosti.
Fant
Mutiran gen
Vrsta genetske bolezni
THE
COL1A1
Avtosomno dominantna
II
COL1A1 in COL1A2
Avtosomno dominantna
III
COL1A1 in COL1A2
Avtosomno dominantna
IV
COL1A1 in COL1A2
Avtosomno dominantna
V.
IFITM5
Avtosomno dominantna
TI
SERPINF1
Avtosomno recesivno
VII
CRTAP
Avtosomno recesivno
VIII
NAJEM 1
Avtosomno recesivno
* Opomba: očitno so mutacije v COL1A1 in COL1A2, ki povzročajo prve štiri oblike nedovršene osteogeneze, genetske spremembe z nekoliko drugačnimi lastnostmi. V nasprotnem primeru ne bi bilo smiselno ločevati enega od drugega.
Simptomi, znaki in zapleti
Vse vrste nepopolne osteogeneze so odgovorne za oslabitev kosti, tako da ima oseba, ki jo bolezen prizadene, posebno nagnjenost k zlomom. Stopnja oslabitve kosti se spreminja glede na obliko; pri nekaterih je to oslabitev večja kot pri drugih.
Ob tem je treba poudariti, da ima vsaka oblika nepopolne osteogeneze svojo simptomatsko sliko, ki se nekaterim lahko spomni simptomatološke slike drugih oblik.
MOŽNI SIMPTOMI IN ZNAKI
Možni simptomi in znaki nepopolne osteogeneze vključujejo:
- Prisotnost malformacij kosti;
- Prisotnost kratkega in majhnega telesa (namenjenega kot prtljažnik);
- Težave s sklepi (npr. Ohlapni sklepi);
- Mišična oslabelost;
- Modra, vijolična ali siva očesna sklera;
- Trikotni obraz;
- Sodna skrinja;
- Morfološke anomalije hrbtenice;
- Zobna krhkost;
- Zmanjšanje ali popolna izguba sluha;
- Težave z dihanjem
- Težave, povezane z odsotnostjo ali pomanjkanjem kolagena tipa 1.
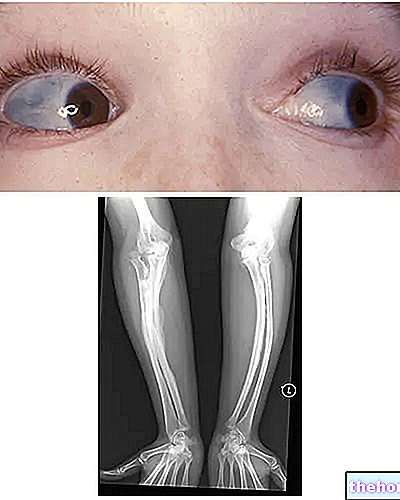
Osteogeneza nepopolna: upoštevajte modro obarvanje beločnic in kostne deformacije, ki so značilne za bolezen. Iz wikipedia.org
KAJ SO NAJZBILJNEJŠE OBLIKE NEPOVOLJNE OSTEOGENEZE?
Zdravniki razvrstijo simptomatološko resnost različnih vrst nepopolne osteogeneze na lestvici 3 stopinj, ki so: blaga stopnja, zmerna stopnja in huda stopnja.
Samo ena oblika spada v kategorijo "blage stopnje": "osteogenesis imperfecta tipa I"; 4 oblike imperfecta osteogenesis imperfecta spadajo v kategorijo "zmerne stopnje": IV, V in VI; nazadnje v kategorijo "hude stopnje" spadajo 3 oblike: II, III, VII in VIII.
TIP I: ZNAČILNOSTI
Najpogostejša in najmanj huda oblika osteogenesis imperfecta tipa I ima naslednje značilnosti:
- Povzroča zlome zlasti pred puberteto;
- Na višino nima "skoraj nobenega vpliva, zato so bolniki običajno normalne" višine;
- Povzroča težave s sklepi in mišično oslabelost
- Odgovoren je za modro, vijolično ali sivo beločnico;
- Je vzrok za trikotne anomalije obraza in hrbtenice;
- Skoraj nikoli ne povzroči deformacije kosti. Če jih izzove, so minimalne;
- Lahko povzroči krhkost zob in / ali izgubo sluha (slednje se običajno pojavi v odrasli dobi);
- Povezan je s prisotnostjo kolagena tipa I, ki je po kakovosti normalen, po količini pa nenormalen (slabši je od običajnega).
TIP II: ZNAČILNOSTI
Osteogeneza imperfekta tipa II je značilna:
- Vzrok smrti ob rojstvu ali kmalu zatem. Težave z dihanjem skoraj vedno povzročijo smrt;
- Prisotnost velike krhkosti kosti in hude kostne deformacije;
- Kratka rast in nerazvita pljuča
- Modra, vijolična ali siva beločnica;
- Prisotnost količinskih in kvalitativnih anomalij kolagena tipa I.
TIP III: ZNAČILNOSTI
Osteogeneza imperfekta tipa III ima naslednje značilnosti:
- Čeprav je zelo resen, ne povzroča pogosto smrti v obdobju novorojenčkov;
- Povezan je z "visoko krhkostjo kosti;
- Odgovoren je za nizko rast, težave s sklepi, mišično oslabelost (zlasti v nogah in rokah), prsni koš, trikotni obraz in nenormalno ukrivljenost hrbtenice;
- To je posledica modre, vijolične ali sive beločnice;
- Lahko povzroči težave z dihanjem, krhkost zob in izgubo sluha;
- Pogosto je odgovoren za deformacije kosti;
- Povezan je s kvalitativnimi in količinskimi nepravilnostmi kolagena tipa I.
TIP IV: ZNAČILNOSTI
Za osteogenezo tipa IV so značilni:
- Stopnja krhkosti kosti med oblikami II in III in obliko I;
- Krajši od povprečne rasti;
- Modra, vijolična ali siva beločnica;
- Kostne deformacije blage / zmerne entitete, rahle nenormalnosti hrbtenice in prsi;
- Trikotni obraz;
- Možna prisotnost krhkosti zob in izguba sluha;
- Prisotnost nenormalnosti kolagena tipa I.
TIP V: ZNAČILNOSTI
Osteogeneza imperfekta tipa V je na nek način podobna osteogenezi imperfekta tipa IV. Vendar pa ima nekaj posebnosti, ki so:
- Sklera normalne barve;
- Odsotnost zobne krhkosti;
- Nastanek nenormalnih kostnih žuljev med procesom celjenja zlomljenih kosti;
- Kalcifikacija medkostne membrane, ki se nahaja med polmerom in ulno. To poslabša gibljivost podlakti.
TIP VI: ZNAČILNOSTI
Tudi osteogenesis imperfecta tipa VI je podobna obliki IV. Za razliko od slednjih je nekaj posebnosti, vključno z visoko koncentracijo alkalne fosfataze v krvi in prisotnostjo na nekaterih kosteh lamel (kostnih), podobnih bodicam rib.
TIP VII: ZNAČILNOSTI
Simptomatsko je lahko osteogeneza imperfekta tipa VII v nekaterih okoliščinah podobna tipu IV, v drugih pa tipu II.
Posebnosti te resne patološke oblike vključujejo:
- Nizka rast;
- Prisotnost izredno kratkega humerusa (kost roke) in stegnenice (stegenska kost);
- Pogosta prisotnost deformacije kolka, znane kot coxa vara.
TIP VIII: ZNAČILNOSTI
Osteogeneza imperfekta tipa VIII zelo spominja na oblike II in III.
Med svojimi posebnostmi izstopajo: hud primanjkljaj rasti, huda skeletna hipomineralizacija in odsotnost (ali redka prisotnost) encima prolil 3-hidroksilaze.
Diagnoza
Na splošno se diagnostični proces, pri katerem so podvrženi bolniki s sumom na obliko osteogeneze imperfecta, začne s skrbnim fizičnim pregledom in natančno anamnezo; nato se nadaljuje z "analizo bolnikove družinske anamneze in z vrsto diagnostičnih slikovnih testov (rentgenski žarki, CT itd.); končno se konča s kvantitativno in kvalitativno oceno kolagena tipa I in z genetski test.
Danes obstaja možnost diagnosticiranja nepopolne osteogeneze tudi v prenatalni fazi, tako da se nosečnica podvrže ultrazvoku.
POMEMBNOST CILJNEGA PREGLEDA IN ZGODOVINE
Zdravnik, strokovnjak za osteogenezo imperfecta, lahko zelo pogosto diagnosticira omenjeno bolezen tudi le s fizičnim pregledom in anamnezo. To pomeni, da ti diagnostični testi niso zanemarljivo pomembni.
VREDNOTENJE PROIZVODNJE KOLAGENA TIPA I
Kvalitativna in kvantitativna ocena kolagena tipa I je praviloma zelo zanesljiv test, saj je, kot je navedeno, za večino primerov nepopolne osteogeneze značilne mutacije v genih, ki nadzorujejo proizvodnjo kolagena tipa 1.
Za oceno količine in kakovosti kolagena tipa I, ki je prisoten na celični ravni pri posamezniku, se lahko zdravniki opirajo na biopsijo kože ali določen krvni test.
Oba ocenjevalna testa sta precej zapletena in bolnik (ali njegovi starši) bodo morda morali počakati nekaj tednov, da bodo vedeli rezultate.
GENETSKI TEST
Z genetskim testom, ki preiskuje celotno DNK preiskovanega posameznika, lahko zdravniki dokončno odločijo o značilnostih prisotne genetske mutacije.
Na splošno je izvedba genetskega testa na vsej celični DNK predvidena, kadar ocena značilnosti kolagena tipa I ni dala želenih rezultatov ali če ne gre za mutacijo v COL1A1 ali COL1A2, ki povzroča "osteogenezo imperfecta.
PRENATALNA DIAGNOSTIKA
Prenatalni ultrazvok je zelo uporaben pri prepoznavanju nepopolnosti osteogeneze tipa II in tipa III.
Terapija
Trenutno ni posebnega zdravila za nedovršeno osteogenezo, z drugimi besedami, ljudem z nepravilno osteogenezo je namenjeno, da živijo z omenjenim stanjem do smrti, kar je pogosto posledica posledic same bolezni.
Pomanjkanje posebne terapije ne izključuje obstoja drugih oblik zdravljenja. Pravzaprav so med terapevtskimi možnostmi bolnika z nepopolno osteogenezo vključene različne simptomatske terapije; s simptomatskimi terapijami mislimo na zdravljenje, ki lahko lajša simptome, upočasni potek bolezni in prepreči (ali vsaj odloži) najhujše posledice.
MOŽNI SIMPTOMATSKI ZDRAVLJENJA
Na seznamu možnih simptomatskih zdravil za osteogenezo imperfecta izstopajo:
- Kirurško vstavljanje nohtov v najdaljše kosti (N.B .: najbolj nagnjeni k zlomom), ki zagotavljajo večjo odpornost na zlome in deformacije. Ta operacija se imenuje rodding intramedularno;
- Konzervativno ali kirurško zdravljenje zlomov in / ali deformacij kosti;
- Zobozdravstvena nega za varovanje zdravja zob;
- Terapije za lajšanje bolečin v primeru zelo bolečih več zlomov;
- Fizioterapija za podaljšanje in krepitev mišic.Elastičen in toničen mišični aparat vam omogoča preprečevanje padcev, ki bi lahko povzročili različne zlome kosti;
- Uporaba pripomočkov za gibanje, vključno z invalidskimi vozički, naramnicami, berglami itd.
PREDNOSTI GIBANJA
Za posameznike z nepopolno osteogenezo zdravniki priporočajo stalno prakso telesne vadbe in gibanja na splošno, saj obe dejavnosti prispevata k krepitvi skeletnega in mišičnega sistema.
Med priporočenimi športi so: plavanje, saj gre za "telesno aktivnost z majhnim vplivom na skeletni sistem", in hoja.
KORISTI ZDRAVEGA ŽIVLJENJA
Vodenje zdravega življenja, izogibanje kajenju, pitju odvečnega alkohola, preveč in slabe prehrane itd. Ima za bolnike z nepopolno osteogenezo več kot diskretne koristi, saj upočasni napredovanje bolezni in zmanjša krhkost kosti.
SIMPTOMATSKE OBRAVNAVE V EKSPERIMENTACIJSKI FAZI
Trenutno zdravniki in raziskovalci ocenjujejo učinkovitost nekaterih simptomatskih zdravljenj, vključno z zdravljenjem z rastnim hormonom in intravensko in peroralno terapijo na osnovi bisfosfonata.
Zaenkrat so rezultati zgoraj omenjenih preiskovalnih zdravljenj dobri za celotno medicinsko skupnost.
Prognoza
Osteogeneza imperfecta je bolezen z negativno prognozo, saj je neozdravljiva, drastično ogroža kakovost življenja in v nekaterih primerih povzroči prezgodnjo smrt prizadete osebe.
Vendar je treba opozoriti, da lahko tudi zahvaljujoč sodobnim simptomatskim zdravljenjem veliko posameznikov z blago obliko osteogeneze imperfecta vodi prijetno in zadovoljivo življenje.
Preprečevanje
Na žalost trenutno ne obstaja preventivni ukrep proti nedovršeni osteogenezi.









-quando-preoccuparsi.jpg)