Splošnost
Izraz pigmentni retinitis (RP) opredeljuje skupino genetskih bolezni, za katere je značilna progresivna degeneracija mrežnice.

Retinitis pigmentosa je distrofija mrežnice, za katero je značilna postopna izguba fotoreceptorjev in disfunkcija pigmentnega epitelija, kar pomeni, da mrežnica postopoma zmanjšuje sposobnost prenosa vizualnih informacij v možgane preko optičnega živca.
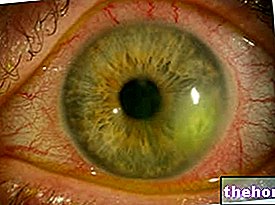
Patološki proces se začne s spremembami pigmentnega epitelija mrežnice. Ko napreduje pigmentni retinitis, pride do redčenja krvnih žil, ki oskrbujejo mrežnico, ki se podvržejo atrofiji. Ob pregledu fundusa se značilno odlagajo vidno. Retinalni pigment ( od tod tudi ime po bolezni). Atrofične spremembe in poškodbe lahko vključujejo tudi optični živec in postopoma odmrejo fotoobčutljive celice mrežnice.
Bolniki, ki jih prizadene retinitis pigmentosa, imajo sprva težave z vidom, zlasti v slabo osvetljenem okolju, in se pritožujejo zaradi zožitve perifernega vidnega polja. Osrednji vid je prihranjen do poznejših stadij bolezni, končni izid pa se lahko dramatično razlikuje: mnogi ljudje s pigmentozo retinitis ohranijo omejen vid skozi vse življenje, drugi pa popolnoma izgubijo vid.
Retinitis pigmentosa je dedna bolezen, ki jo večinoma povzročajo genetske spremembe, ki jih prenašajo eden ali oba starša. Vrsta genetske okvare določa, katere retinalne celice so najbolj vpletene v motnjo, in s kliničnega vidika omogočajo razlikovanje med različnimi stanji. Do danes je bilo ugotovljenih več kot 50 različnih genetskih napak, povezanih s pigmentozo retinitisa. Nenormalnosti se lahko prenesejo od staršev na potomce po enem od treh vzorcev dedovanja: avtosomno recesivno, avtosomno dominantno ali heterosomalno recesivno (vezano na X ali X).
Simptomi
Za dodatne informacije: Simptomi retinitisa v pigmentozi
Retinitis pigmentosa običajno najdemo pri mladostnikih in mladih odraslih. Simptomi se pogosto pojavijo med 10. in 30. letom starosti, vendar je diagnozo mogoče postaviti v zgodnjem otroštvu ali veliko kasneje v življenju.
Zgodnji simptomi pigmentnega retinitisa lahko vključujejo:
- Težave pri videnju ponoči (nočna slepota) ali pri slabi svetlobi
- Počasno prilagajanje iz vida v temi v svetlobo in obratno;
- Zoženje vidnega polja in izguba perifernega vida;
- Občutljivost na svetlobo in bleščanje.
Nekateri simptomi so odvisni od vrste fotoreceptorjev. Palice so odgovorne za črno -bel vid, medtem ko stožci omogočajo razlikovanje barv.
V večini primerov pigmentnega retinitisa najprej prizadenejo palice. Vendar pa se lahko v hitro razvijajočih oblikah stožci prizadenejo tudi v zgodnji fazi.
Palice so koncentrirane v zunanjih delih mrežnice in se aktivirajo pri šibki svetlobi, zato njihova degeneracija vpliva na periferni in nočni vid. Če gre za stožce, lahko pride do izgube zaznavanja barv in osrednjega vida.
Prevladujoči delež fotoreceptorjev določa posebna napaka v pacientovi genetski sestavi.
Pogosto je prvi simptom pigmentoznega retinitisa nočna slepota (ali noktalopija). Nekateri menijo, da potrebujejo vedno več časa, da se prilagodijo razlikam v svetlobi, ko se premaknejo iz dobro osvetljenega območja v temnejše. Tipična oblika izgube vida povzroči zožitev perifernega vida (tunelski ali teleskopski vid); ta vzorec se imenuje obročni skotom. Včasih ta pojav v zgodnjih fazah morda manjka, opazimo pa ga, ko se posameznik pogosto spotakne ob predmete ali je vpleten v prometno nesrečo. Kadar izguba vida vključuje osrednje območje mrežnice (imenovano tudi makularna distrofija) težave pri branju in podrobnem delu, ki zahteva koncentracijo na en sam predmet, na primer previjanje niti skozi ušesce igle Mnogi bolniki poročajo, da so videli bliskavice svetlobe (fotopsijo), ki so pogosto opisane kot majhne, utripajoče in utripajoče luči.
Stopnja napredovanja bolezni in stopnja izgube vida se od osebe do osebe razlikujejo. Nekateri ekstremni primeri se lahko hitro razvijejo v dveh desetletjih, drugi počasno, ki nikoli ne vodi do popolne slepote. Zgodnji začetek se pojavi pri hujših oblikah pigmentnega retinitisa, medtem ko se pri bolnikih z blažjimi stanji (npr. Avtosomno dominantno) lahko bolezen razvije v petem ali šestem desetletju življenja. kot ženske in huje; samice po drugi strani prenašajo genetsko lastnost (nosijo spremenjeni gen na kromosomu X) in manj pogosto kažejo simptome motnje.
Zapleti
Retinitis pigmentosa bo še naprej napredoval, čeprav počasi. Vendar je popolna slepota redka, lahko pa pride do znatnega zmanjšanja perifernega in centralnega vida.
Pri bolnikih s pigmentozo retinitisa se v zgodnji starosti pogosto pojavi otekanje mrežnice (makularni edem) ali katarakta. Te zaplete je mogoče zdraviti, če motijo vid.
Sorodne bolezni
Običajno bolnik s pigmentozo retinitis nima drugih motenj in v tem primeru govorimo o "nesindromski" ali preprosti pigmentozi retinitisa. Vendar imajo nekateri sindromi nekatere klinične simptome pri tej očesni bolezni; najpogostejši je Usherjev sindrom, ki prizadene približno 10-30% vseh bolnikov s pigmentozo retinitisa in je povezan s sočasno prirojeno ali progresivno izgubo sluha. Pri Leberjevi prirojeni amaurozi pa lahko otroci v prvih šestih mesecih življenja oslepijo ali skoraj oslepijo. Druge bolezni, povezane s pigmentozo retinitisa, vključujejo Bardet-Biedlov sindrom in Refsumovo bolezen.
Vzroki
Bolezen lahko povzročijo številne genetske okvare: pravzaprav obstaja več genov, ki lahko, če jih prizadene sprememba, povzročijo fenotip retinitis pigmentosa. Ti običajno kodirajo beljakovine, vključene v transdukcijsko kaskado, ki omogoča vid, vpliva na celično transkripcijo (ki pošiljajo napačna sporočila celicam mrežnice) ali za elemente, ki sestavljajo strukturo fotoreceptorjev. Podedovane genske mutacije so v celicah prisotne od trenutka spočetja; pogoste nenormalnosti vključujejo gene RP1 (pri retinitis pigmentosa-1, avtosomno dominantni) , RHO (RP4, avtosomno prevladujoč) in RDS (RP7, avtosomno prevladujoč). Ne dedni vzroki za pigmentozo retinitisa so redki, vendar obstaja možnost odkritja osamljenega primera (spontana mutacija), pri katerem ni družinske anamneze. bolezni.